人类基因组测序的技术进步
陈浩源:University of Georgia 生化博士, Emory University 生化博士后。 具有DNA分析和健康相关的软件开发经验,熟悉多种定量分析和统计分析方法 :熟悉基因克隆、蛋白表达、纯化、蛋白质特性、酶催化机制,传统方式研究酶-底物间的相互作用(如预稳态动力学、稳态动力学,化学修饰),以及分子生物学技术(定点突变技术)。
(版权所有,转载请联系作者)
十万个为什么 3.0丛书
为什么首次人类基因组测序需要多国合作完成,而现在能够在临床诊断中完成患者的基因组测序?
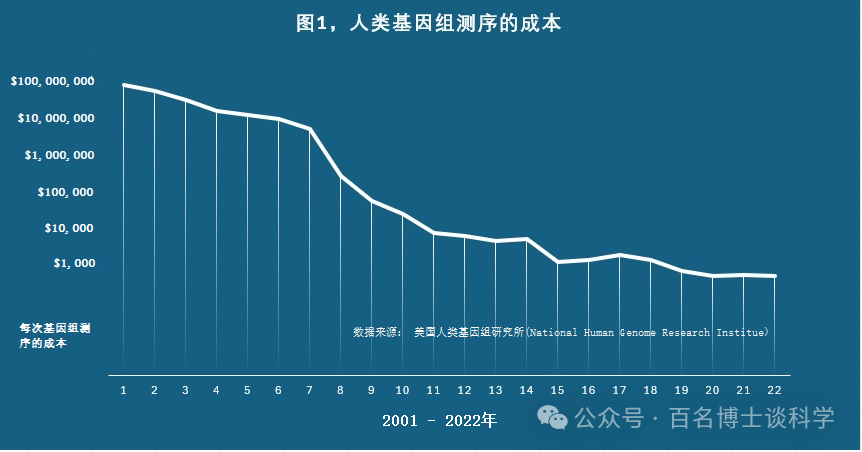
首次人类基因组测序(人类基因组计划)是一项国际科学研究项目,其目标是确定构成人类 DNA 的碱基对,并从物理和功能角度对人类基因组的所有基因进行识别、绘图和测序。 它于1990年开始,并于2003年完成。美国、英国、法国、德国、日本和中国科学家共同参与了这一预算达30亿美元的人类基因组计划。随着人类基因组逐渐被破译, 使人类第一次在分子水平上全面地认识自我。科学家通过研究遗传密码,深入了解基因如何相互作用以及与环境相互作用。此外,他们还可了解这些相互作用如何影响生物体的发育和健康。科学家使用人类基因组数据来研究疾病或医疗状况,识别和评测药物靶点并开发新的治疗方法,以及筛选和测试潜在的药物。由于基因组数据据有以上的巨大应用,催动了一系列基因组测序的技术革命,大大的降低了基因组测序的成本,特别是开发了高通量测序技术,也称为下一代NGS测序(Next Generation Sequencing) ,将人类基因组测序的成本降至约少于1,000美元,将测序时间缩减到几个小时,使基因测序技术能够应用在临床中诊断患者的基因组(图1)。以下通过比较人类基因组计划中使用的传统的测序方法和高通量的NGS测序方法,说明在过去的二十年中,NGS技术的发展取得的显著进步 (如图2)。
1. 测序样品准备:
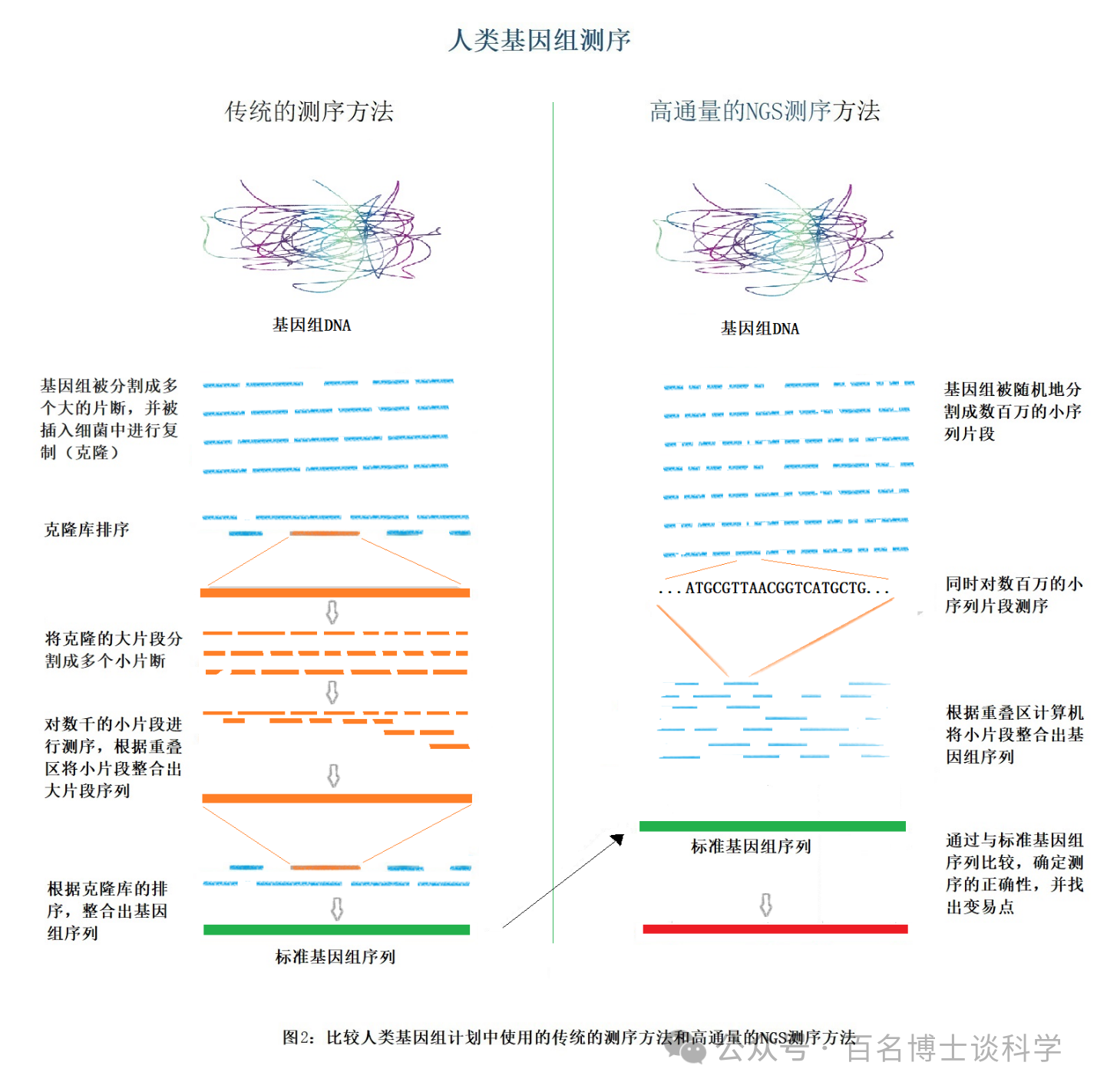
人类基因组由23对染色体组成,含有约31.6亿个DNA碱基对,其中一部分的碱基对组成了大约20000到25000个基因(也就是能够制造蛋白质的编码序列),全部基因只占总长度的约1.5%。由于基因组非常庞大,因此需要把大分子DNA随机地“敲碎”成许多小片段,收集这些随机小片段并将它们全部连接到合适的测序载体。小片段测序完成后,根据重叠区计算机将小片段整合出大分子DNA序列。这就是所谓的鸟枪测序法。
在人类基因组计划中,由于当时的测序仪无法同时测序多个DNA小片段,因此提出了“分级鸟枪测序法”:基因组被分割成多个片断(长度接近150,000个碱基对),在确定这些片断的相对顺序后,将这些片断插入细菌中,并利用细菌进行复制(克隆),以获得大量的纯化的片断,通过对每一个这样的片断分别应用“鸟枪测序法”,最终将这些片断通过配对末端法,以及其他许多定位数据重新组装在一起从而获得完整的基因组。在“分级鸟枪测序法”中, 工作量最大的部分,应该是如何确定克隆库中DNA片断的相对顺序。通常要构建各种限制性酶切图谱,并参考遗传连锁图谱,cDNA图谱所揭示的基因相对位置。
在高通量NSG测序中,测序样品的准备要简单的多,基因组DNA直接被分割成数百万的小序列片段。许多NSG测序仪要求PCR扩增, 基于单分子测序法的NSG测序仪甚至可直接对小序列片段进行测序,而不必扩增DNA片段以增强信号。这样可以减少样品制备的成本和时间,并降低了扩增过程中产生的偏差和错误。
2. 基因测序
传统的测序的一般策略是进行 4 个独立的反应,各产生一套放射性标记的单链寡核苷酸,它们有固定的起点,另一端终止于不同的 A、T、G 或 C 位点。每个反应的产物在高分离度的聚丙烯酰胺凝胶上电泳分级。 经放射自显影,DNA 序列可从凝胶上直接读出。 其中最常用的是双脱氧测定法(Sanger 法),双脱氧法或酶法利用DNA聚合酶合成单链DNA模板的互补拷贝,DNA聚合酶能在退火于“模板”DNA的引物3‘ 端上进行链的延伸(如图3)。通过与模板碱基的特异性配对,脱氧核糖核苷酸(dATP, dCTP, dGTP and dTTP)被掺入到引物的生长链上。链的延伸是通过引物生长端的3‘羟基与被掺入脱氧核糖核苷酸的5‘磷酸基的反应形成磷酸二酯键,在总体上看,链是从 5‘ →3‘ 方向延伸的。双脱氧测序法利用了DNA聚合酶也能用双脱氧核糖核苷酸(ddATP, ddCTP, ddGTP and ddTTP)为底物的特性。当ddNTP被掺入到延伸着的引物的3‘ 端时,由于链上3‘ 羟基的缺如,链的延伸就终止于G、A、T或C。在 4 个测序反应中,每个反应只需各加入 4 种可能的ddNTP 中的一种,就将产生如图 3 所示的 4 个序列阶梯。调整每个测反应中的ddNTP 与dNTP的比例,使引物的延伸在对应于模板DNA上的每个可能掺入ddNTP的位置都有可能发生终止。
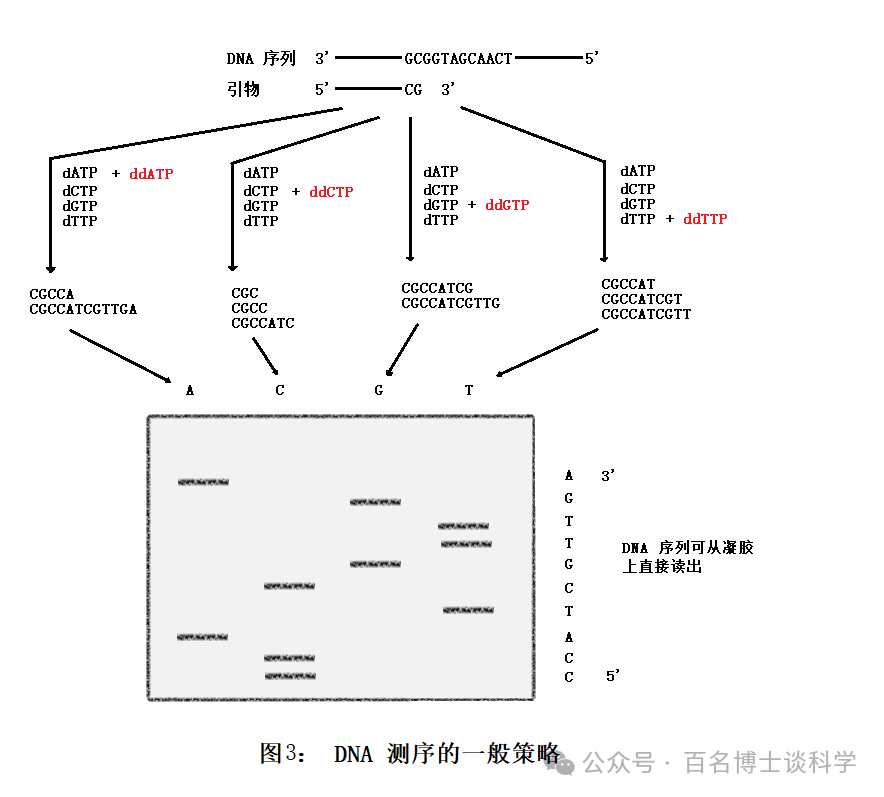
这样的测序方法,每次可大概测定2千个碱基对。人类基因组大约包含31亿个碱基对,这意味着需要作致少一百五十万次测序。这就是为什么首次人类基因组测序需要多国合作,历时十多年才完成。
最流行的NGS测序技术是基于 “短序列片段测序”,虽然短序列片段NGS只能处理1,000 碱基对左右的DNA序列,但该测序法可以并行处理数百万到数十亿次序列片段。在短短几个小时内,NGS可以测序到Gbytes级别的数据。这一技术显著增加了基因组测序的吞吐量,并使在临床诊断和治疗中使用NGS成为可能。不同的测序仪公司推出的NGS测序技术有所不同,在NGS测序领域的著名公司有罗氏,Illumina,Ion Torrent和Applied Biosystems等,他们的NGS测序技术在他们的网站都有详细的介绍。以下是罗氏的NGS测序法,这种方法中,DNA被分割成较短的序列片段,不多于1,000 碱基对。将这些DNA片段固定在链霉包被的磁性小颗粒上,每个颗粒上具有一个特定DNA片段。然后通过PCR扩增DNA片段,在单个颗粒上产生数百万份拷贝。然后,将颗粒分布在刻蚀过的玻璃芯片的单孔中,用于测序。在测序期间,依次加入四种脱氧核苷酸,即dATP,dCTP,dGTP和dTTP,与DNA序列片段反应。当核苷酸添加到DNA序列片段时,会释放出生物光信号。光信号的强度被记录下来,并转化成DNA片段的序列信息。每次反应后,剩余的脱氧核苷酸会被洗去,并接着加入下一种脱氧核苷酸进行反应。通过循环加入四种脱氧核苷酸进行反应,来读取DNA 片段的序列。
3. 测序数据分析
采用适宜的序列分析方法和软件,对得到的核酸测序下机数据进行序列拼接,最终获得待测微生物样本的全基因组序列信息。传统的测序方法对DNA片段逐个进行测序,因此拼接相对容易;而NGS测序数据的分析则比较复杂。一般包括以下部骤:
A. 测序反应的评估
这个步骤主要由测序仪提供的软件完成,给出测序反应的程度,质量等指标。例如读出量(Yield),错误率等。
B. 清除低质量的测序数据
低质量的测序数据通常包括那些太短的读出片段,因为这些短的读出片段在拼接中不起太大作用。此外太长的读出片段也会被删除,因为这些长的读出片段通常错误率也高。这个步骤还包括去除重复的读出片段,DNA片段PCR放大后会产生大量的重复的读出片段。通过比较重复的读出片段可排除错误的测序数据。
用于这个步骤的软件有很多,比较常用的有FASTQC。通常还会根据具体的需要作一些软件开发。
C. 序列拼接
人类基因组计划以经为我们提供了标准的基因组序列,理论上可以通过将读出片段直接与标准基因组序列比照,拼接出基因组序列。但是以下情况使这种方法需要太高的计算成本:1)标准的基因组序列太长(含有约31.6亿个DNA碱基对);2)读出片段太短 (50 - 150DNA碱基对),导致不准确和低效率;3)由于存在基因变易,基因组序列与标准的基因组序列并不完全一样。针对以上情况,已经开发出各种软件,比较好的有BAW和Bowtie2,这类软件有如下特点:
a) 将基因组序列排列成前缀树(prefix trie),减少对共同序列(前缀)的对比次数。
b) 利用压缩字符的Burrows-Wheeler算法来减少前缀树对内存的需求。
c) 对前缀树进行回溯获得所有的子序列。
d) 允许不精确匹配,使存在变易的片段仍能比较。
D. 变易分析
比较基因组序列与标准的基因组序列,找出变易的部分可以说是基因组测序的目的。根据不同的目的,以经开发了各种软件,例如,GATK, SAMtools和Mpileup等。